家庭診療/兒童結締組織或骨的遺傳性疾病
| 醫學電子書 >> 《默克家庭診療手冊》 >> 兒童保健 >> 兒童骨骼肌肉系統疾病 >> 兒童結締組織或骨的遺傳性疾病 |
| 默克家庭診療手冊 |
|
|
|
某些影響結締組織的疾病是遺傳性的。這些疾病包括埃-當氏綜合征、馬方綜合征、彈性假黃瘤、皮膚松弛癥和粘多糖病。骨軟骨發育不良影響骨或軟骨,骨硬化病影響骨骼。
埃-當氏綜合征
埃-當氏(Ehlers-Danlos)綜合征是一種非常罕見的遺傳性結締組織疾病,臨床主要特征為關節過度靈活、皮膚彈性過強和組織脆弱。
埃-當氏綜合征本身有幾種變性,是由那些控制形成結締組織的各種基因變異所致。多數患兒僅有十分靈活的關節(良性的運動過度),而無其他癥狀。經過一段時間后,患兒關節的過度靈活性可減輕。
目錄 |
癥狀和診斷
皮膚可捏起10余cm,但是釋放后能恢復到正常位置。關節可特別靈活。在身體的骨表面經常易形成廣泛的傷痕,特別是在肘部、膝部和脛骨部。皮下可形成小而堅硬的圓形結節,這種結節可在X線片上顯現。
輕度的碰撞可以引起寬闊的裂傷,并通常伴有少量出血。少數埃-當氏綜合征患者有容易出血的傾向。縫線往往撕裂脆弱的結締組織,因而增加了修補傷口的難度。身體內部器官也同樣脆弱,也使手術變得困難。扭傷和脫位極為常見。患兒中,大略25%的逐漸出現脊柱異常彎曲(脊柱后側凸)所致的駝背,并且90%的為扁平足。疝氣和腸道憩室也極為常見。很少有胃腸道出血或破裂(穿孔)。
患此綜合征的孕婦,可因其身體組織容易伸展而導致早產。如果胎兒已患此綜合征,羊膜可能出現早破。此外由于結締組織脆弱,采用剖腹產術或陰道口切開術(外陰切開術)來幫助嬰兒娩出,可能會存在更多的困難。分娩前、分娩期或分娩后可能會發生嚴重出血。
預后和治療
盡管患埃-當氏綜合征會發生多種多樣的并發癥,但多數患者都具有正常的壽命。對于少數患者,并發癥(如血管破裂)卻會致命。
目前,尚無治愈該病的方法。由于組織的脆性,患者應避免受到外傷。經常穿用代保護性的衣服和墊子會有益處。如果埃-當氏綜合征患者希望有小孩,應作遺傳咨詢,以確定嬰兒可能被遺傳的風險程度。
馬方綜合征
馬方(Marfan)綜合征是一種罕見的,導致眼、骨骼、心臟和血管異常的遺傳性結締組織疾病。
癥狀
馬方綜合征的遺傳基因是顯性的(見遺傳學),但并非每一位遺傳有此基因者均會患此病。患者的身高超過了他們的年齡和家庭成員。手臂間距(雙臂從體側平伸時手指間的距離)超過自身身高,手指細長。胸骨常有外凸或內陷畸形。關節極為靈活。扁平足和由于脊柱異常彎曲(脊柱后側凸)的駝背,疝氣等常見。通常,皮下脂肪少,上腭高。
眼晶體脫位。醫生利用檢眼鏡,通過瞳孔即可看見晶體脫位的邊緣。患者還可能有高度近視和視網膜(眼后的感光區)剝離。
可因主動脈壁層薄弱而逐漸引起重要的動脈擴張,形成動脈瘤(一種血管壁的凸出)(見主動脈瘤和夾層動脈瘤)。血液可滲入血管壁層之間(壁間動脈瘤),或動脈瘤破裂,導致大出血。隨著主動脈擴張,導致心臟與主動脈交界處的主動脈瓣膜閉鎖不全(動脈返流)(見心臟瓣膜疾病)。位于左心房和左心室間的二尖瓣,可能出現閉鎖不全或脫垂(向后凸出進入左心房)。肺部可能出現充滿積液的囊腫(見胸膜疾病)。囊腫可能破裂而引起氣胸(空氣進入胸膜腔),導致呼吸困難。
并發癥的危險性主要取決于畸形的嚴重程度。主要的危險是主動脈突然破裂,導致迅速死亡。破裂很可能發生在運動期間。
診斷與治療
如果一個異乎尋常地高、消瘦的人有上述典型癥狀的任何一種,醫生可以懷疑其患馬方綜合征。然而,許多的該綜合征患者可以不出現任何癥狀和異常。
治療的主要目的是預防血管和眼睛的疾病。應每年檢查一次眼睛。如果視力出現問題,應該立即就診。
利血平或普萘洛爾可用來減輕血流對管壁施加的壓力,預防主動脈擴張和破裂。如果主動脈已出現擴張,有時需要手術修補或置換受損部位。
患馬方綜合征的兒童身材往往趨向于長得很高。醫生可以使用激素(雌激素和孕酮)來治療長得太高的女孩。這種治療通常可促使青春期在10歲時早熟,從而使其停止生長。
馬方綜合征患者若希望生育應該尋求遺傳咨詢,以確定他們的孩子被遺傳馬方綜合征的危險程度。
彈力纖維性假黃瘤
彈力纖維性假黃瘤是一種遺傳性結締組織疾病,主要影響皮膚、眼睛和血管。
彈力纖維性假黃瘤主要影響彈性纖維。使頸部、腋下、腹股溝和臍周的皮膚增厚,起皺紋、無彈性和松弛,微黃的鵝卵石樣花紋結節使皮膚像一種毛小雞外觀。視網膜的改變可能導致視力的嚴重損害,甚至失明。動脈可能變窄,導致胸痛和間歇性跛行(因為血液供給不足,導致活動時出現腿痛)。可能發生鼻出血和腦部、子宮及腸道的出血。高血壓常見。
預后與疾病和動脈受損的嚴重程度有關。患者通常在30~70歲期間死于并發癥。
皮膚松弛癥
皮膚松弛癥是一種罕有的皮膚極易延伸和松弛下垂的結締組織性疾病。
皮膚松弛癥主要影響彈性纖維。本癥病因不清楚,通常是遺傳性的,但常無家族史。有些病例遺傳特征相當輕微,有些導致智力遲鈍,而其他的一些可能是致命的。
癥狀
在遺傳型,皮膚可能在出生時就非常松弛或出生后變得松弛。皮膚松弛在面部特別顯著,所以患兒面容呈悲哀狀或“丘吉爾”貌。典型的鉤狀鼻子。常有疝氣和腸道憩室。肺氣腫可能導致肺動脈高壓癥,出現輕微的肺心病。
非遺傳型(獲得型)皮膚松弛癥的臨床表現與遺傳型有所不同。它發病較晚,并影響皮膚以外的其他部位,受累的皮膚看起來與遺傳型皮膚不同,而且鼻子不是鉤狀的。特別在兒童或青少年,皮膚松弛癥發病類似一些嚴重疾病,伴發燒、廣泛的身體漿膜炎和嚴重的皮疹(多形性紅斑)。在成人,疾病可能不知不覺地發生,有時因肺部并發癥或主動脈破裂導致死亡。
診斷和治療
對皮膚松弛癥,通過檢查皮膚極易確診。
皮膚松弛癥尚不能治愈。對遺傳型患者,整形外科能夠成功地改善外貌,但是,皮膚可能再次變松弛。整形外科對獲得型的治療少有成功。同樣需要治療肺部疾病,如肺氣腫或肺動脈高壓癥。
粘多糖病
粘多糖病是一種以典型面部特征和骨骼、眼睛、肝臟以及脾臟畸形的遺傳性疾病,有時伴隨智力障礙。
這類疾病的根本原因是身體儲存過多的粘多糖使體內粘多糖含量過高。粘多糖是結締組織的主要成分。同樣,過剩的粘多糖進入血液并且堆積于全身,部分隨尿液排泄。粘多糖有許多不同的類型,需要不同的生化降解酶。所以,在許多粘多糖病的類型中,每一種特殊的遺傳缺陷影響一種特殊的生化酶。
癥狀和診斷
通常,粘多糖病患兒在出生時沒有癥狀,到嬰兒期和兒童期,身材矮小、骨骼發育不良、多毛以及身體發育不良才逐漸明顯。面容可能已經丑陋、厚嘴唇、張口以及鞍型鼻。依據粘多糖病的類型,受累的患兒有些有正常的智力,而多數患兒在出生時智力似乎正常,幾年后卻逐漸地顯現出智力遲鈍。少數類型的患兒,有眼睛受累,出現角膜渾濁和視力障礙。其家庭成員也可能有粘多糖病。
醫生根據癥狀可以作出初步診斷。粘多糖病可以通過羊水或絨毛膜取樣(見遺傳疾病檢查診斷),進行分離和篩查異常酶活性,在患兒出生前作出診斷。出生后,采集尿樣品亦可篩查。但是,尿檢查結果常常不準確,因此,必須進行血液和其他檢查才能確定診斷。X線片上顯示的骨骼畸形對診斷有幫助。
預后和治療
預后依賴于粘多糖病的類型。某些類型可引起早期死亡。
目前,該病尚無治療方法。置換異常酶的設想作用是有限的,或僅可獲得暫時的成功。骨髓移植(見移植)對本病作用不大,而且,經常引起死亡或殘疾。因此,對該病的治療仍有爭論。
骨軟骨發育不良
骨軟骨發育不良是一組骨或軟骨生長異常的遺傳性疾病。
許多類型的骨軟骨發育不良均可引起身材矮小(侏儒)。軟骨發育不良是各類肢體短小侏儒癥中最常見的類型之一。該病發生率約在25000~40000活產嬰兒中出現1例,男女和所有人種均可發病。
癥狀
每一種不同類型有不同的癥狀,例如,軟骨發育不良的癥狀是肢體短小、膝內翻、大額、鞍形鼻以及駝背。
駝背
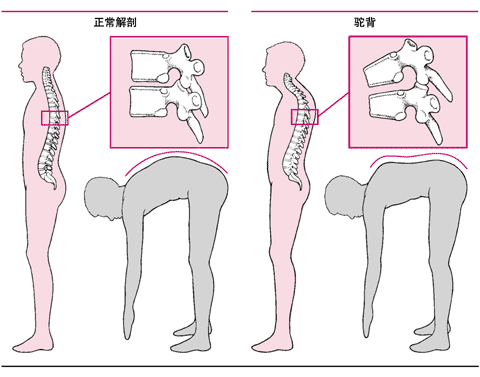
骨骼的疾病,如髖關節脫位是所有骨軟骨發育不良的典型癥狀。在少數類型,第二椎骨的畸形(齒狀突發育不良)使第二椎骨從第一椎骨脫位并且壓迫脊髓,可立即導致死亡。
診斷和治療
因為每一種類型由不同的遺傳變異引起,其遺傳性和病程不同,預后也不同,醫生應盡力去獲取準確的診斷。可在嬰兒出生以前進行確診。在一些病例可直接通過光纖鏡(胎產鏡)或超聲掃描觀察胎兒。假如一個骨軟骨發育不良患者或其近親希望要一個孩子,就得冒遺傳因素影響孩子的危險。
當關節疾病嚴重影響患兒功能時,有時需施行人工關節置換術以替代這個受累關節。第二椎骨畸形必須給予手術固定,以預防脊髓損傷。
骨硬化病
骨硬化病(大理石樣骨)是一種骨密度增加和骨骼成形異常的遺傳性疾病。
骨硬化病在有些病人幾乎不引起殘疾,而另一些則為進行性的并且是致命的。一些類型沒有明顯的癥狀(遲發型);在另一些類型,癥狀在幼年即開始顯現(早發型)。
大部分類型無特殊的治療。必須外科手術緩解顱骨畸形所致的顱內高壓。徹底切開骨周圍神經,可以抑制骨骼的過度生長,如果這個過度生長的骨壓迫神經,需要外科手術來減輕壓迫。牙移植,可以預防特有的牙咬合異常(錯合);牙正畸治療可以糾正這些情況。骨的過度生長可能引起面部嚴重扭曲,有時會導致心理障礙。
遲發型
癥狀可出現在兒童期、少年和青年期。大理石樣骨病(阿爾貝斯.舍恩貝格病)是遲發型中輕微的一種,可以沒有癥狀。這種類型分布相當廣泛,可出現在任何國家和任何人種中。通常,患兒在出生時骨骼正常,至兒童期骨密度增加。確診常常帶有偶然性,如在X線攝片中無意發現骨密度增高。患者面容、體格、智力和壽命正常,身體健康。有時,由于骨的過度生長壓迫神經造成面癱或耳聾。可逐漸出現輕微的貧血。
早發型
早發型骨硬化病較少見,但對嬰兒卻是一種潛在的致命性疾病。癥狀包括生長發育不良和體重不增,容易擦傷、異常出血以及貧血。肝臟和脾臟腫大,眼神經和面神經受損。患兒常在1歲內由于貧血、嚴重感染或出血而死亡。有些患兒X線片顯示出骨密度增加,并隨時間而加重。骨髓移植在有些患兒已獲得成功,但是遠期預后尚不清楚。
幼年變形性骨軟骨病
骨軟骨病是一組影響兒童發育期骨骺(出現于發育期不同的骨化中心)的疾病,結果為異常的骨生長和骨畸形。骨軟骨病的病因尚不清楚。不同的疾病影響的骨骼不同,受侵襲的骨在股骨頭骨軟骨炎是股骨;在脛骨結節骨骺壞死癥是脛骨;在脊柱骨軟骨病是脊椎骨,以及在足舟骨骨軟骨病是一種足的小骨(足舟骨)。
股骨頭骨軟骨炎
股骨頭骨軟骨炎(萊-卡-珀氏病)是股骨頭的骨骺破壞。這種疾病在5~10歲的小孩中發生率為1/1000~1/5000,常見于男孩。疾病通常僅影響單側髖關節,造成股骨頭的供血不足,但是供血不足的原因不明。
主要癥狀是髖關節疼痛和行走困難。癥狀逐漸開始且進展緩慢。關節活動受限,大腿肌肉因缺乏活動而萎縮。在X線片上,股骨頭開始變扁平;稍后,股骨頭出現斷裂。
治療包括長期臥床休息、牽引、懸吊、石膏褲和夾板。一些專家建議使用整形外科來矯正髖關節的所有殘余畸形。
該病不經治療,可在2~3年后自愈。如果該病留有后遺癥,在髖關節會增加并發骨關節炎的危險。如果疾病得到恰當治療,則很少發生骨關節炎。
脛骨結節骨骺壞死癥
脛骨結節骨骺壞死癥(奧-施氏病)是在脛骨結節骨骺和軟骨的炎癥。
本病多發于10~15歲的男孩。該病的病因據認為是由于附著于脛骨粗隆下端的髕韌帶過度牽拉所致的損傷。這個脛骨粗隆下端稱為脛骨結節。
該病通常影響單側脛骨。主要癥狀是在脛骨粗隆髕韌帶附著部位的疼痛、腫脹和觸痛。X線的膝關節片上可發現脛骨粗隆裂斷。
唯一的治療措施是用止痛劑減輕疼痛和限制有關的運動及過度的鍛煉,尤其是避免膝關節過度彎曲。癥狀通常在病程幾周或幾月之后自行消失。有時候,必須用石膏模固定腿。在某些病例,可以局部注射氫化可的松,手術切除骨質碎片,骨鉆孔引流減壓直到康復,或者進行骨移植。
脊柱骨軟骨病
脊柱骨軟骨病是一種由于脊椎變型引起的背痛和駝背。
這種病多發于青少年,男孩較女孩多見。脊柱骨軟骨病或許不是一種單一的疾病,而是具有相似特征的一組疾病。病因目前尚不清楚。
通常,輕型病例是在學齡兒童常規篩查脊柱畸形時才被檢查出來。癥狀包括整個上背部的持續的輕度疼痛。脊柱上部的彎曲大于正常彎曲度。該病病程可持續一個較長時期,甚至達幾年,但是癥狀輕微。癥狀消失后脊柱可能遺留輕微的彎曲。
輕度非進行性病例可通過減輕負重,減小背部張力,以及避免劇烈的活動來治療。偶爾,駝背較嚴重時,必須采用脊柱支架或者睡硬板床。若是病情發展,有時候需用外科手術矯正。
足舟骨骨軟骨炎
足舟骨骨軟骨炎(克勒骨病)是一種罕見的足舟骨和軟骨的炎癥(骨軟骨炎)。
該病常見于3~5歲的男孩。癥狀有患足腫脹、疼痛及觸痛,尤其是在足弓的內側。在足部負重或者行走時疼痛加重。常有跛行。該病病程往往為數月但是很少超過2年。止痛和避免足負重可減輕癥狀。倘若在早期階段足底對足弓形進行恰當的支撐,膝下進行允許行走的石膏固定數周,對疾病可能有效。
| 關于“家庭診療/兒童結締組織或骨的遺傳性疾病”的留言: | |
|
目前暫無留言 | |
| 添加留言 | |